- 原文链接:Multi-omics integration reveals functional signatures of gut microbiome in atherosclerosis
- 发表时间:2025年12月
摘要
本研究整合了动脉粥样硬化相关的多队列肠道微生物组和宿主转录组数据,通过批次校正、差异分析、Meta分析、多样性分析、功能推断、代谢通路推断、代谢物-宿主基因关联分析以及随机森林建模,系统挖掘了与动脉粥样硬化相关的微生物功能特征。结果发现,AS患者的肠道菌群组成和功能均发生明显变化,筛得 5 个稳定变化的菌属、2 个关键代谢物(H₂O₂ 和 Ethanol)以及 2 个宿主基因(GPX2 和 FANCD2),并构建了潜在的“微生物-代谢物-宿主基因”关联网络。此外,这 5 个菌属在多种验证策略下表现出较好的诊断性能和一定特异性,提示其可作为AS非侵入性生物标志物的候选。该研究展示了多组学整合分析在复杂疾病机制解析和生物标志物筛选中的实用价值。
一、文章简介
简单说,作者想搞清楚肠道菌群是怎么影响动脉粥样硬化(AS)的,完全利用公共数据,通过一系列分析和验证,最后找出5个菌属 + 2个代谢物 + 2个宿主基因,构成了和动脉粥样硬化相关的“微生物-代谢物-宿主基因”三联关系;进一步利用随机森林模型和交叉验证,筛选出5个菌属作为AS诊断标志物。
数据来源: 全部来自公共数据库,没做新实验:
- 肠道菌群:6个数据集,宏基因组+16S,共567个样本
- 宿主基因:8个数据集,RNA-Seq+芯片,共420个样本
- 代谢信息: MetaCyc 和 STITCH 数据库
分析方法: 差异分析、MetaDE、批次校正(ConQuR、Combat)、多样性分析(alpha/beta diversity、PCA/PCoA)、PRMT代谢推断、leave-one-genus-out菌群贡献分析、DM score宿主效应推断、随机森林建模、交叉验证(5-fold CV、LOSO、study-to-study transfer)。

二、主要结论
1. AS 与对照的菌群结构差异
在做差异分析之前,先用 PCA/PCoA 看不同队列之间的数据是否“混在一起”或“分得太开”。结果显示,经过批次效应校正后,数据可比性更好了;同时,AS 和对照组在 Alpha 多样性、Beta 多样性上都存在明显差异。并通过 Meta 分析整合多个队列后,作者找到了大量差异菌,其中有 6 个菌属在宏基因组和 16S 数据中变化一致。
fig2:

2:推断代谢功能(图S8A-B)
利用 MetaCyc 数据库 和 PRMT 算法,从差异微生物基因(DEMGs)出发,从差异微生物基因 → MetaCyc数据库 → PRMT算代谢物积累/消耗趋势 → LOGO分析,预测了微生物群落对53种代谢物的“消耗”或“积累”趋势。LOGO分析 量化了单个菌属对特定代谢物(如 H₂O₂ 和 Ethanol)的贡献度,建立了菌属与代谢物的功能联系。
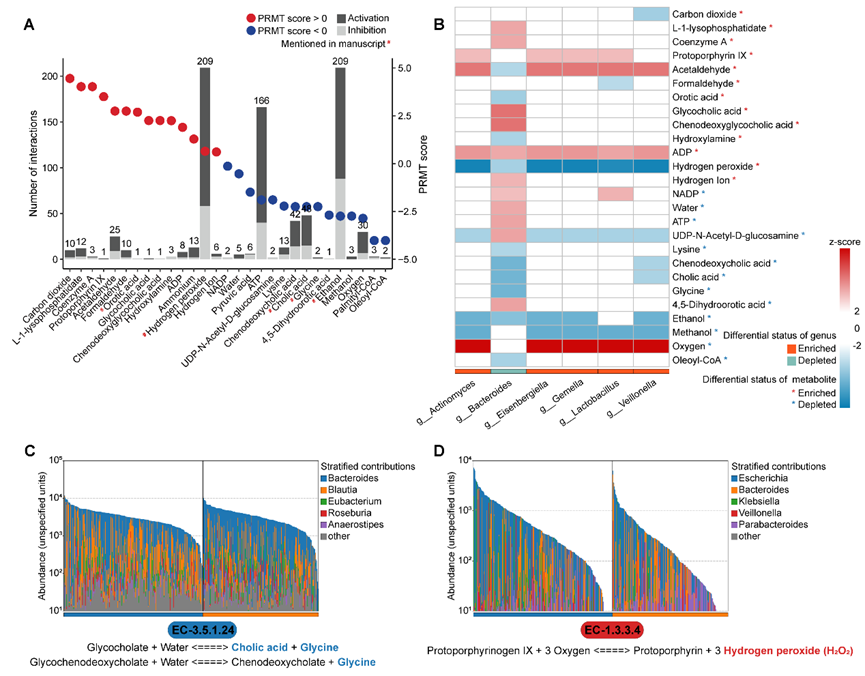
3. 搭起“菌-代谢物-基因”完整链条(图3A-B)
这是本文最核心的套路。作者首先从 STITCH 数据库中提取代谢物与宿主基因的互作信息。为了将微生物端的“信号”与宿主端的“响应”联系起来,他们提出了一个关键约束:微生物预测的代谢物变化趋势(PRMT评分)必须与代谢物对宿主基因的疾病影响(DM评分) 方向一致。最终,成功构建了5条“菌属(如 Bacteroides)—代谢物(如 H₂O₂)—宿主基因(如 GPX2)”的完整互作链条,这就把“菌”和“人”真正串起来了,形成一个完整的致病/保护假说。

4:文献佐证生信分析结果
查已有文献,看筛出来的菌、代谢物、基因是否曾被报道与AS相关。这也是文章的一个亮点,生信分析最怕被人说“算法硬凑出来的”。这一操作是告诉审稿人我的结果不是凭空冒出来的,是有文献背景的。既有新发现,又不脱离已有认知。
作者以表格形式展示文献调研结果:
| Type | Node/Edge | Type of evidence | Evidence (PMID) |
|---|---|---|---|
| Genus | Actinomyces | Indirect | 39262241 |
| Bacteroides | Direct Direct Indirect Indirect |
32989686 29018189 30571343 31726978 |
|
| Eisenbergiella | - | - | |
| Gemella | Indirect | 34869642 | |
| Veillonella | Direct Indirect Indirect |
37220524 31726978 31812509 |
|
| Metabolite | Ethanol | Direct | 30885430 |
| Direct | 37286970 | ||
| Direct | 11067787 | ||
| Direct | 24582196 | ||
| Direct | 31906033 | ||
| H2O2 | Direct Direct |
16009356 32245238 |
|
| Host gene | FANCD2 | Indirect | 37689092 |
| GPX2 | Indirect | 20848490 | |
| Genus-Metabolite | Actinomyces – Ethanol | - | - |
| Bacteroides – H2O2 | Direct | 24164536 | |
| Eisenbergiella – Ethanol | - | - | |
| Gemella – Ethanol | - | - | |
| Veillonella – Ethanol | - | - | |
| Metabolite-Host gene | Ethanol – FANCD2 | Indirect | 21919919 |
| H2O2 – GPX2 | Indirect | 25261240 |
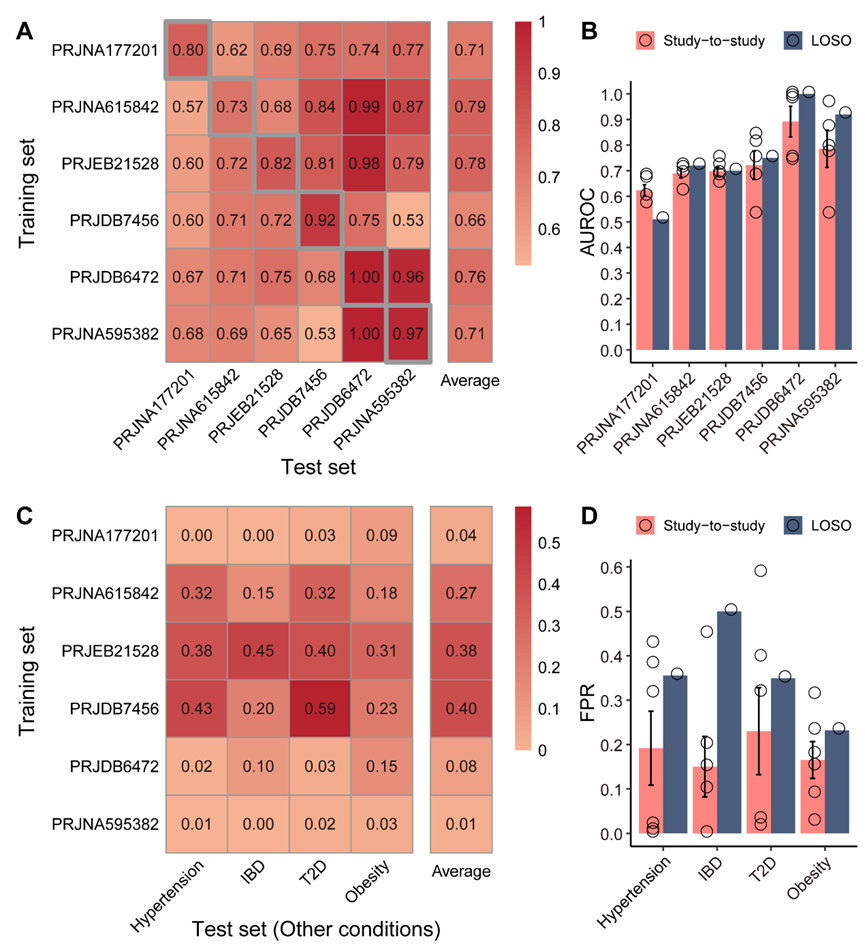
4:验证5个菌属作为AS非侵入性诊断标志物的潜力
作者选出 5 个菌属作为候选 biomarker,用 Random Forest 建模,并进行了5-fold CV、LOSO、study-to-study transfer三种交叉验证 (s11A–B)和特异性验证(s11C–D)
三、文章小结
这篇论文展示了一条非常清晰的公共数据多组学整合分析套路,纯生信分析的高分文章,文章分析过程和思路简单,但搜集整理数据的难度和工作量较大:
- 收集公开数据、文献
- 批次效应校正
- 差异丰度分析和Meta分析
- 菌群多样性和群落结构
- 推断功能和代谢潜力
- 连接代谢物和宿主基因
- 诊断模型标志物建模和验证